Junge Lab
Harald Junge, Ph.D.
Mary and Marian Robinson Chair in Macular Degeneration
Associate Professor
Department of Ophthalmology and Visual Neuroscience
University of Minnesota
Room 310A LRB, 2001 6th St SE
Minneapolis, MN 55455
email: [email protected]
Research Topics
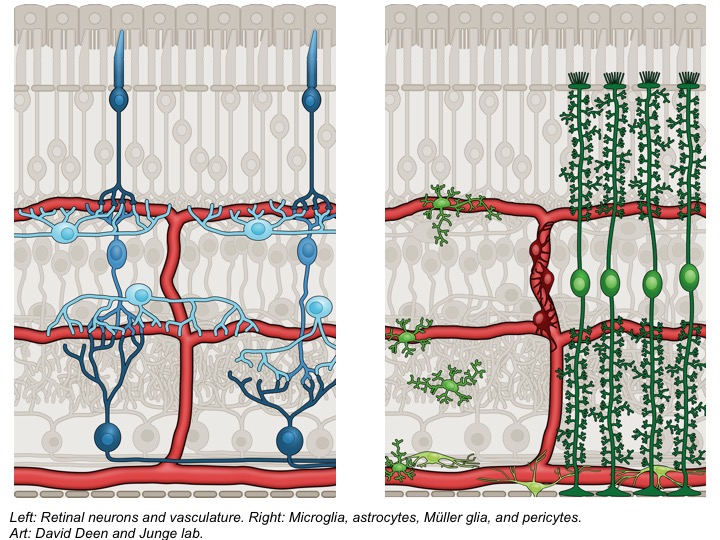
- Neurovascular interactions
- Endothelial cell-pericyte interactions
- Norrin and Wnt signaling
- Frizzled4/LRP5 and Frizzled4/LRP6 agonists
- Blood-retina barrier, blood-brain barrier
- Retinal angiogenesis
- Cystoid macular edema in AMD, DR
- Familial exudative vitreoretinopathy (FEVR)
- Small vessel disease
Overview
The major goals of my laboratory are to understand cell-cell signaling impinging on the CNS vasculature and use that knowledge to target pathways for pharmacological intervention. Areas of focus are regulation of the blood-retina barrier and blood brain barrier by neural signals, endothelial cell – pericyte interactions and CNS angiogenesis. We seek to understand how signaling pathways function at the molecular level, create genetic mouse models of disease, and test novel drug candidates for therapeutic intervention. We use integrated approaches including mouse genetics, cell-based assays, molecular manipulations (e.g., CRISPR/Cas9), scRNA-Seq, and proteomics to understand processes at molecular, cellular, developmental, and disease levels.
Significance
Neurovascular communication is critical for the function of the retina and brain. For instance, neural signals induce and maintain the blood-CNS barriers (including blood-brain and inner blood-retina barrier). These barriers limit non-selective diffusion of solutes from the blood into the CNS. They also mediate highly
selective transport of nutrients, proteins and hormones into the CNS, and of waste products out of the CNS. Blood-CNS barrier dysfunction is implicated in prevalent retinal diseases, such as age-related macular degeneration (AMD) and diabetic retinopathy (DR). Barrier dysfunction is also implicated in neurological diseases, for
example stroke, neurodegenerative diseases, and epilepsy. A direct consequence of impaired barrier function is cystoid macular edema in several prevalent retinal diseases. Therefore, it is important to understand the mechanistic basis of barrier maintenance and how the barriers can be pharmacologically modulated by interfering with neurovascular signaling.
The appropriate formation of CNS blood vessels by outgrowth from pre-existing vessels (angiogenesis) occurs in development and is essential for retina and brain function. Once an appropriate vasculature has been formed, blood vessels become quiescent and cease to form additional ramifications of the CNS vascular tree. However, in pathological conditions, blood vessels may become reactivated and form super-numerous and aberrant neo-vessels that damage the surrounding tissue and cause edema. In ocular diseases, including neovascular AMD and proliferative DR, pathological growth of neo-vessels leads to impaired vision or blindness. Anti-angiogenic therapies that target the VEGF
proangiogenic signal are not always effective or cease to be effective during long-term treatment. This may be in part because CNS angiogenesis is governed by specialized neural signals besides VEGF. Therefore, it is important to understand CNS-specific mechanisms of angiogenesis, including the role of wnt and norrin signaling.
Pericytes are functional components of the neurovascular unit and play pivotal roles in maintaining the integrity and functionality of the CNS vasculature. These mural cells ensheathe capillaries, contribute to blood-brain barrier function, modulate developmental angiogenesis and vessel stabilization, contribute to CNS fibrotic scar formation, modulate cerebral blood flow (predominantly at the transition of arterioles to capillaries), and influence immune and inflammatory responses. Loss or dysfunction of pericytes is associated with neurological disease, including DR, ischemic stroke, small vessel disease, and neurodegenerative disease. However, there is a lack of efficacious
therapeutic approaches that target pericytes or endothelial cell- pericyte interactions. Our research into mechanisms of endothelial cell – pericyte interactions as well as preclinical research using novel pharmacological interventions seeks to address this unmet medical need.
Progress
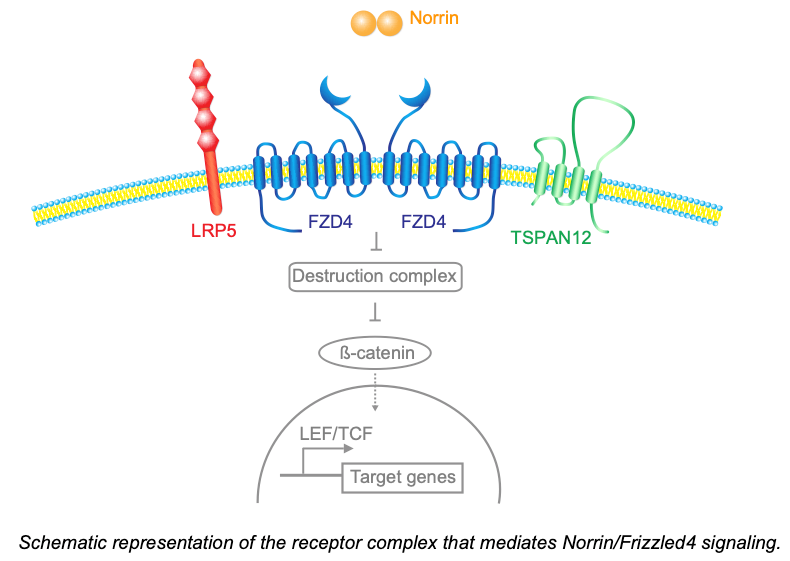
A major focus of our studies on neurovascular signaling is the norrin/frizzled4 pathway, which is essential for CNS angiogenesis and blood-CNS barrier function. Congenitally impaired norrin/frizzled4 signaling causes familial exudative vitreoretinopathy (FEVR), or X-linked norrie disease in male infants. This pathway is best understood in the retina, although it also has functions in other CNS regions, for example the cerebellum. The ligand Norrin is released from the neuroretina (predominantly müller glia and horizontal cells) and activates signaling in vascular endothelial cells via a receptor complex containing Frizzled4 and LRP5 or LRP6. Frizzled4 engages other membrane proteins to form receptor complexes in a combinatorial manner. This enables Frizzled4 to respond selectively to distinct ligands. We reported the first of such receptor complex co-factors, the tetraspanin family member Tspan12 (Junge et al. 2009) and showed that Tspan12 is a co-receptor for Norrin that increases the ligand selectivity of Frizzled4 towards Norrin (Lai et al., 2017). This work explained why mutations in Tspan12 cause familial exudative vitreoretinopathy.
We created a mouse model that is highly useful to understand the role of blood-CNS barrier defects in retinal disease and test pharmacological interventions. Inducible deletion of the Tspan12 gene in vascular endothelial cells — after developmental angiogenesis is complete — creates a disease model characterized by blood-retina barrier defects in an otherwise largely intact vasculature (Zhang et al. 2018). This model showed that blood-retina barrier dysfunction directly results in cystoid edema in mice.
In collaboration with the University of Toronto and a biotech startup, AntlerA, we found that Frizzled4/LRP5 agonists activate beta-catenin dependent signaling in bEnd.3 brain endothelial cells in vitro and in mice in vivo, thus providing the first demonstration of in vivo efficacy of this novel class of drug agonists. We used Tspan12 KO mice and endothelial cell-specific conditional Tspan12 gene disrupted mice, which are models of retinopathy due to impaired norrin/frizzled4 signaling, and showed that systemic or topical Frizzled4/LRP5 agonist antibodies restore norrin/frizzled4 signaling (Chidiac and Abedin et al. 2021, Zhang et al. 2023). ScRNA-seq of Tspan12 KO mice with and without pharmacological restoration of norrin/frizzled4 signaling provided a detailed insight on how norrin/frizzled4 signaling limits unspecific transport and enables specific transport at the blood-retina barrier (Zhang et al., 2023). We found that Frizzled4/LRP5 agonist alleviate blood-retina barrier dysfunction and cystoid edema in our mouse models, and this finding translated well in subsequent AMARONE clinical trials conducted by EyeBio (now Merck). These preclinical and clinical data suggest that Frizzled4/LRP5 agonists may become a novel treatment option for patients with AMD and DR.

Outlook and research platform
Current projects focus on the regulation of the blood-retina barrier and blood brain barrier, which may lead to a better understanding how CNS edema can be controlled, for example cystoid macular edema. Current basic science studies include the role of the MDM2-P53 node in controlling norrin/frizzled4 signaling, the role of endothelial calcium in control of norrin/frizzled4 signaling, and the role of actin binding proteins as mediators of norrin/frizzled4 signaling in angiogenesis and blood-retina barrier function. Explorative projects include studies in the area of age-related macular degeneration. Translational projects include testing
Frizzled4/LRP5 and Frizzled4/LRP6 agonists for restoration of pericyte coverage in the retina and brain.
Selected Publications
A Frizzled4-LRP5 agonist promotes blood-retina barrier function by inducing a Norrin-like transcriptional response. Zhang L, Abedin M, Jo HN, Levey J, Dinh QC, Chen Z, Angers S, Junge HJ. iScience. 2023 Aug 18;26(8):107415.
A Norrin/Wnt surrogate antibody stimulates endothelial cell barrier function and rescues retinopathy. Chidiac R, Abedin M, Macleod G, Yang A, Thibeault PE, Blazer LL, Adams JJ, Zhang L, Roehrich H, JoHN, Seshagiri S, Sidhu SS, Junge HJ, Angers S. EMBO Mol Med. 2021
Jul 7;13(7):e13977. doi: 10.15252/emmm.202113977. Epub 2021 Jun 9. PubMed PMID: 34105895; PubMed Central PMCID: PMC8261507.
Endothelial Cell-Specific Inactivation of TSPAN12 (Tetraspanin 12) Reveals Pathological Consequences of Barrier Defects in an Otherwise Intact Vasculature. Zhang C, Lai MB, Pedler MG, Johnson
V, Adams RH, Petrash JM, Chen Z, Junge HJ. Arterioscler Thromb Vasc Biol. 2018 Nov;38(11):2691-2705.
Ligand-Selective Wnt Receptor Complexes in CNS Blood Vessels: RECK and GPR124 Plugged In. Junge HJ. Neuron. 2017 Aug 30;95(5):983-985.
TSPAN12 is a Norrin Co-Receptor that Amplifies Frizzled4 Ligand Selectivity and Signaling. Lai MB, Zhang C, Shi J, Johnson V, Khandan L, McVey J, Klymkowsky MW, Chen Z, Junge HJ. Cell Rep. 2017 Jun 27;19(13):2809-2822.
Norrin-induced Frizzled4 endocytosis and endo-lysosomal trafficking control retinal angiogenesis and barrier function. Zhang C, Lai MB, Khandan L, Lee LA, Chen Z, Junge HJ. Nat Commun. 2017 Jul 4;8:16050. doi: 10.1038/ncomms16050.
TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Junge, HJ, Yang, S, Burton, JB, Paes, K, Shu, X, French, DM, Costa, M, Rice, DS, and Ye, W. Cell, 139(2):299-311. 2009.
Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Junge, HJ, Rhee, J, Jahn, O, Varoqueaux, F, Spiess, J, Waxham, MN, Rosenmund, C, and Brose, N. Cell, 118(3):389-401. 2004.
See complete publication list here:
www.ncbi.nlm.nih.gov/sites/myncbi/1byHor9eEgkAK/bibliography/42260874/public/?sort=date&direction