Sun lays out a cellular timeline for the rise and spread of cancer
The timelines of life are many and varied: the timeline of life on Earth, the timeline of plants from seeds to germination, the timeline of gestation from conception to birth, and the timeline of a lifespan.
Cancer can affect the timeline of a human life. Thanks to advances in genome sequencing and machine learning, cancer cells have revealed an evolutionary timeline from their earliest mutations to the emergence of a dominant tumor clone among subclones, an evolutionary journey that typically takes years. Knowledge of molecular and cellular timelines is valuable for developing targeted therapies for many diseases, cancer among them.
 As we reported three years ago, LMP assistant professor Ruping Sun and his colleagues pinpointed “a key divergence in the relentless logic of cancer’s spread,” the evolutionary “branching” of the tumor from its seedbed of primary cancer cells over time. In Sun’s hands, cells become molecular clocks. Differences in their genomic sequences serve as timestamps on a cell’s journey from a normal state to a mutated one to a malignant clone with a growth advantage to a dominant tumor clone among subclones, a “founder” cancer cell. Founder clones possess their own unique set of DNA mutations and alterations of chromosomes, the threadlike gene-bearing bodies in the cell nucleus. The “clock-like behavior” of the process is revealed through elegant learning algorithms and computing power, as described by Sun and his coauthors in “Evolving copy number gains promote tumor expansion and bolster mutational diversification” published in Nature Communications in March.
As we reported three years ago, LMP assistant professor Ruping Sun and his colleagues pinpointed “a key divergence in the relentless logic of cancer’s spread,” the evolutionary “branching” of the tumor from its seedbed of primary cancer cells over time. In Sun’s hands, cells become molecular clocks. Differences in their genomic sequences serve as timestamps on a cell’s journey from a normal state to a mutated one to a malignant clone with a growth advantage to a dominant tumor clone among subclones, a “founder” cancer cell. Founder clones possess their own unique set of DNA mutations and alterations of chromosomes, the threadlike gene-bearing bodies in the cell nucleus. The “clock-like behavior” of the process is revealed through elegant learning algorithms and computing power, as described by Sun and his coauthors in “Evolving copy number gains promote tumor expansion and bolster mutational diversification” published in Nature Communications in March.
"By integrating the history of genomic variants with the dynamics of tumor expansion, we provide new insights into using copy number gains for early cancer detection and targeted therapy,” Sun said in an interview.
Copy number gains – what the researchers term somatic copy number alterations (SCNAs) -- refer to additional chromosomes or chromosomal segments that cells generate as they move along the timeline from a normal state to the establishment of a tumor founder cell, which Sun and his colleagues term the Most Recent Common Ancestor or MRCA of sequenced tumor samples. From there a dominant founder clone emerges giving rise to a continuously evolving population, leading to the spread of cancer.
Most tumors, especially solid tissue tumors like those of bone, breast, colon, liver, and lung, show extra chromosomes or chromosomal segments upon microscopic and molecular analysis. “We use the terms ‘driver’ and ‘passenger’ to classify chromosomal gains because driver alterations are oncogenic and passenger alterations are ‘along for the ride’ but do not cause cancer,” Sun said. “You can use the terms to describe the functionality of these gains.”
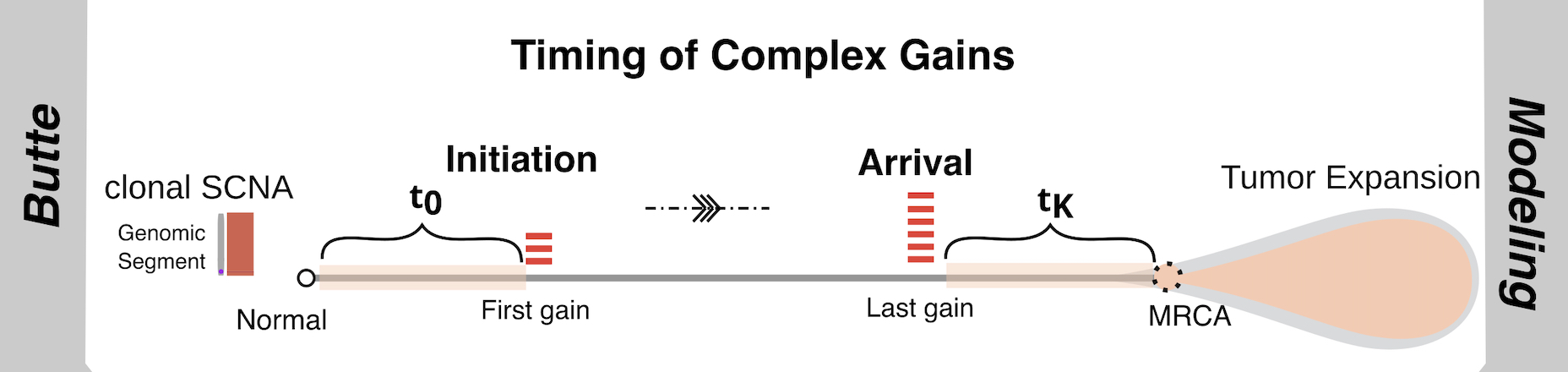
To find out when these alterations arise on the healthy-cell-to-tumor-cell timeline, the investigators created and implemented a machine learning program called BUTTE (BoUnds of Time Till Expansion), a framework they employed to estimate the timing of clonal alterations associated with multiple gains of genetic material, all based on genomic sequence information from cells in the tumor tissue.
Sun and most of his coauthors of the Nature Communications paper are affiliated with the Masonic Cancer Center (MCC), where he is a faculty member in MCC’s Genetic Mechanisms Program. His coauthors include LMP’s postdoc Zicheng Wang, research scientist Yunong Xia, research associate Athanasios Nikolakopoulos, and professor Scott Dehm, a co-leader of MCC's Prostate and Urologic Cancer Translational Working Group. Another coauthor is Yale University assistant professor Jason Sheltzer, lead author of a paper published last year in Science, of which Sun is a coauthor, about how extra chromosomes—long a mystery in tumors—may help them grow and also make them vulnerable to drug therapy.
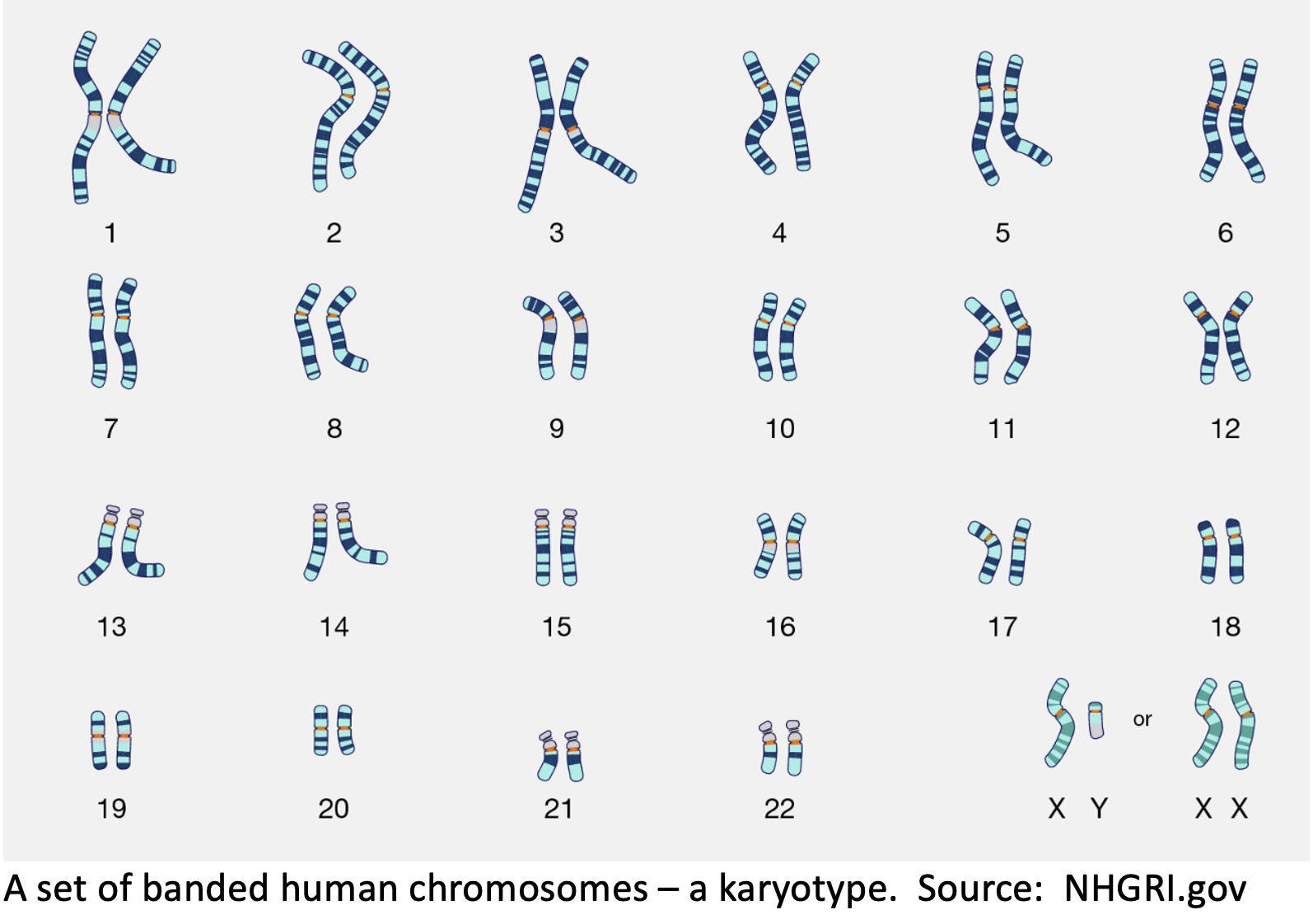
Now a personal note. In the 1980s I worked for LMP professor Jorge Yunis who pioneered high-resolution chromosome banding. When chromosomes are subjected to certain stains, they display light and dark horizontal bands, with the light bands considered to be rich in genes. Yunis’s technique yielded more than a thousand bands in a chromosome set, or karyotype, compared to 320 bands using the standard technique. Chromosome bands help cytogeneticists to spot chromosomal breakage, segment deletions, and translocations in which chromosome arms are exchanged, often a feature of blood cancer cells. Duplications of whole chromosomes or chromosome segments, commonly seen in solid tissue tumors, are also easier to spot.
Evidence of duplicated or missing chromosomes or chromosomal segments is called aneuploidy. In the 1980s no one knew how or when aneuploidy arose or what effect it had on tumor initiation, progression, or metastasis. The Sun team's whole genome sequencing (WGS) analysis using its creative algorithmic tool BUTTE serves as a “lens” on the timeline when cells undergo changes that can result in what Sun calls "chromosomal instability" that in turn, over time, can lead to aneuploidy, malignancy, and metastatic cancer. And all without a pathologist peering into a microscope to discern the banding pattern of chromosomes and see whether they constitute an intact set or whether instability has scrambled them and produced deletions and duplications that can disrupt the normal functions of the cell.
Sun’s team analyzed multiple tumor tissue samples from the same patients with bone, breast, colon, esophagus, and prostate cancer to accurately measure the clonal locations on the timeline. “If we take just one sample via microdissection, the sample may involve only a subclone of that tumor, which may not indicate the clonal mutations in the entire tumor,” he said. “To track tumor progression, you want to have samples along the way."
The rise of aneuploidy may be associated with the tumor microenvironment: the cells, molecules, and structures such as blood vessels that surround and support other cells and tissues. “If there are extra forces in the microenvironment, they may put some selective pressure on tumor cells and you should see that in the evolution – in the genome space,” Sun said. “Actually, by looking at gene sequences inside these gains, we found some enrichment signals for genes participating in proliferative signaling, escaping immune response, and in vascular formation in the tumor.” Tumor angiogenesis is the proliferation of blood vessels penetrating the cancerous growth for the supply of nutrients and oxygen. It is generally required for tumor proliferation and metastasis.
Sun thinks aneuploidy and copy number evolution are “reflective of the microenvironmental stresses” and plans to investigate that hypothesis in follow-up research. He and his team also plan to explore the possibility, reported by other researchers, that chromosomal instability may induce inflammation in the microenvironment that could facilitate the migration and invasiveness of tumor cells.
The rapid advance of DNA sequencing technologies coupled with machine learning algorithms like BUTTE are poised to turn disease evolution timelines into progressively longer timelines for disease-free survival. “I think the field of computational biology is going to shed a lot of light on things we’ve always wanted to know but haven’t had the tools to investigate, until now,” Sun said.



